Osteosarcoma or osteogenic sarcoma is the main type of bone cancer. Osteosarcoma is a type of cancer in which tumour cells produce immature bone known as osteoid. Although osteosarcoma is rare, it is the most common childhood bone cancer. Osteosarcoma usually affects the body's long bones near the growth plates, areas where new tissue forms as a young person grow. Most tumours occur around the knee in either the femur or shin bone. Osteosarcoma can also arise from other parts of the body even outside of the bones in the soft tissues, especially in older patients. More males than females get this cancer.



Overview
What are the most common types of Bone Cancer?
There are several types of bone tumours. They are named according to the area of bone or tissue where they start and the type of cells they contain. The most commonly found types of primary bone cancer are:
- Osteosarcoma
-
- Chondrosarcoma
-
Chondrosarcoma is cancer of cartilage cells. More than 40% of adult bone cancer is chondrosarcoma, making it the most prevalent bone cancer in adults. The average age of diagnosis is 51, and 70% of cases are in patients over 40. Chondrosarcoma tends to be diagnosed at an early stage and often is low grade. Many chondrosarcoma tumours are benign (not cancer). Tumours can develop anywhere in the body where there is cartilage, especially the pelvis, leg, or arm.
- Ewing's Sarcoma
-
Ewing's sarcoma is the second most prevalent type of bone cancer in children and adolescents, and the third most often found in adults. It accounts for about 8% of bone cancers in adults. Ewing's sarcoma can start in bones, tissues or organs, especially the pelvis, chest wall, legs, or arms.
- Chordomas
-
A chordoma is a rare type of cancerous tumour that can occur anywhere along the central portions of the spine, from the skull base to the tailbone. Chordomas are slow-growing tumours, which have bony and soft tissue components. They are notorious to recur after treatment, and in about 20-30 percent of cases, cancer spreads to other areas of the body, such as the lungs or bones. Approximately half of all chordomas occur at the base of the spine (sacrum), about one third occur in the base of the skull (occiput), and the rest occur in the cervical (neck), thoracic (upper back), or lumbar (lower back) vertebrae of the spine. As the chordoma grows, it puts pressure on the adjacent areas of the brain or spinal cord, leading to the signs and symptoms. A chordoma anywhere along the spine may cause pain, weakness, or numbness in the back, arms, or legs. Chordomas typically occur in adults between ages 40 and 70. About 5 percent of chordomas are diagnosed in children. For unclear reasons, males are affected about twice as often as females.
- How common are Chordomas?
-
Chordomas are rare, occurring in approximately 1 per million individuals each year. Chordomas comprise fewer than 1 percent of tumours affecting the brain and spinal cord.
- What are the causes of Chordoma?
-
Changes in the TBXT gene have been associated with chordoma. An inherited duplication of the TBXT gene identified in a few families is associated with an increased risk of developing a chordoma. Duplications and increases in expression of the TBXT gene both result in the production of excess brachyury protein. Brachyury expressed by the tumour cells can be easily be identified by immunohistochemistry.
- Secondary (or metastatic) bone cancer
-
Secondary (or metastatic) bone cancer is cancer that spreads to the bone from another part of the body. This type of bone cancer is more prevalent than primary bone cancer.
- How are primary bone sarcomas classified?
-
Following is the WHO classification of primary bone sarcomas.
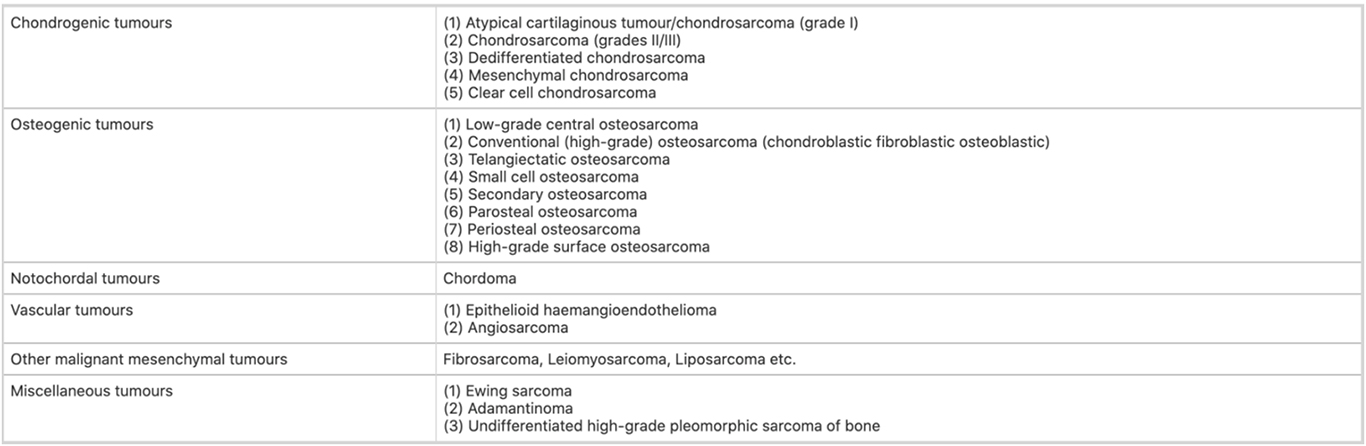
- How are bone sarcomas staged?
-
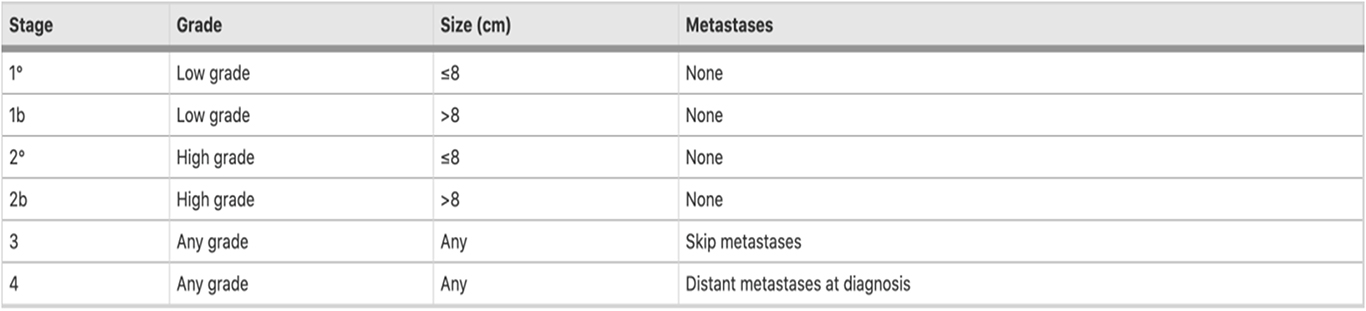
All bone sarcomas are staged as per the following AJCC/TNM staging system.
Symptoms
Patients with a bone tumour will often experience one or more of the following:
- Pain in the area of the tumour is generally described as dull and aching. It may worsen at night and increase with activity.
- Fever and night sweats
- Many patients will not have any symptoms but will note a painless mass instead.
- Although bone tumours are not caused by trauma, an injury can sometimes cause a tumour to start hurting. Injury can also cause a bone that is weakened by a tumour to fracture or break. This may be severely painful.
- Occasionally, tumours may be discovered incidentally when an x-ray is taken for another reason, such as a sprained ankle or knee injury.
Risk Factors
Most bone tumour patients do not have any known risk factors however there is a range of risk factors for bone cancer, including genetics. People with long term inflammatory diseases, such as Paget’s disease, are at an increased risk.
Other risk factors for developing bone cancer include:
- Being under 20 years of age
- Exposure to radiation, such as receiving radiation therapy for a different cancer
- A previous bone marrow transplant recipient
- Having a close relative with bone cancer
- Individuals with hereditary retinoblastoma, a type of eye cancer that most commonly develops in children.
Diagnosis
At specialized centres such as APCC, patients are subjected to a thorough evaluation by the doctor which is followed by one or more of the combination of tools that are used to diagnose bone tumours:
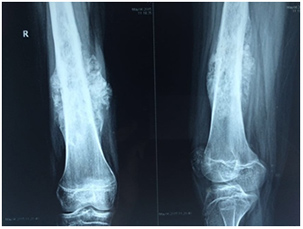
- X-ray is often the first investigations. Sometimes special views are ordered to enable better visualization of bone tumours.
- A biopsy can either be a needle biopsy or a surgical biopsy. Many considerations need to be kept in mind before a biopsy is performed from a bone tumour, especially cancerous tumours. A Multidisciplinary tumour board discusses the best approach to obtain a biopsy when required at APCC. Poorly planned or executed bone biopsy may compromise further treatments.
- CT scans are usually used to help form an initial bone cancer diagnosis and to see whether cancer has spread to other areas of the body. CT scans may also be used to guide the biopsy needle.
- Bone Scan - A radionuclide bone scan may be used to diagnose and stage bone cancer. This bone cancer detection tool may reveal whether the primary tumour has spread to other places in the bone, and how much damage it has caused. In a bone scan, a small dose of radioactive material is injected into a blood vessel, where it travels through the bloodstream. The material then gathers in the bones and is detected by a scanner through nuclear imaging. This test is very sensitive and may find small metastases before they would appear on a regular X-ray. However, other conditions such as arthritis or infection look similar on the scan, so a confirmatory biopsy is often needed.
- MRI may help outline a tumour in the bone and may also help determine whether cancer cells have spread to the brain or spinal cord. It is also used to assess treatment response and is used during follow-up. MRI is probably the best investigation when it comes to spinal tumours.
- PET-CT scan is probably the most sensitive identification tool for cancers. It is an important tool to perform pre-treatment staging of the disease as well as post-treatment monitoring.
- Other tests such as blood workup, Alkaline Phosphatase, ESR, bone-marrow cytology or biopsy, etc are also performed.
- Pathology including immunohistochemistry is crucial for every patient of bone sarcomas.
- Genetic and molecular studies such as EWS-FLI1 translocation for Ewing’s sarcomas and other sarcomas are extremely crucial for a diagnosis.
Treatment
- MDT for Bone Tumours
-
As well as having care delivered or supervised by a specialist bone sarcoma MDT, patients should be allocated a key worker. Children, teenagers and young adults should also be discussed at the relevant children’s or TYA (young adult) MDT. This requires sufficient specialist staff to ensure age-appropriate care. A bone sarcoma MDT should be properly constituted, adhering to the requirements for core membership of the relevant specialties, and meeting minimum criteria for the number of patients treated each year; they should collect data on patients, tumours, treatment and outcomes as agreed.
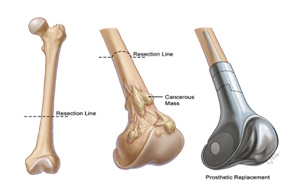
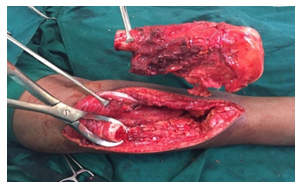
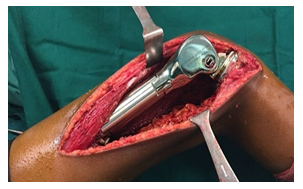
- Surgery for Bone Sarcomas & Limb Salvage Surgery
-
Decisions about the optimal surgical procedure for the primary tumour (i.e. limb salvage or amputation) require MDT discussion, considering tumour size and involvement of anatomical structures, response to neoadjuvant therapies and patient preference. Surgical reconstruction may be influenced by patient and surgeon choice and should follow the open discussion of the risks and benefits of available options and expected functional outcomes.
Surgery for bone sarcomas proceeds in 3 steps.
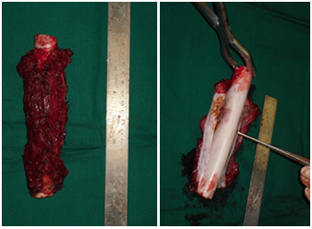
- Resection of the Tumour with adequate margins.
- Reconstruction of bone defect created due to removal of the Tumour.
- Reconstruction of the soft tissue and soft tissue balancing allowing for an early return to function.
Curative surgery aims to resect the whole tumour with adequate margins. Where possible, wide en-bloc resection of the affected part of the bone and involved soft tissue should be performed. Close surgical margins may be marked with (MRI-inert) clips placed in the surgical field. In bone sarcomas, surgery should involve removal of all anatomical structures involved in the original prechemotherapy tumour volume where feasible. The specimen should be orientated to allow the pathologist to describe the anatomical location and thickness of surgical margins.
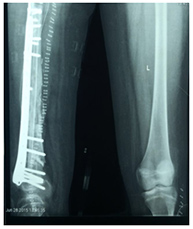
Reconstruction of bone following removal of the Tumour may be biological or non-biological depending on the location of the Tumour and age of the patients. Non-biological reconstructions involve the use of Tumourmegaprosthesis which will allow for an immediate return to function after the surgery. Biological reconstruction involves using allograft bones or autograft, wherein the bone can be removed, sterilized of Tumour cells by giving a single fraction of high dose radiation and reimplantation back into place after stabilizing with suitable implants.
- Systemic Therapy (Chemotherapy/Targeted Therapy)
-
Osteosarcoma
Osteogenic Sarcoma is one of the most common bone tumours and they have a high risk for spreading into the lungs. Hence, along with local therapy like resection, there is also a need for chemotherapy to prevent distant metastases. They are considered chemoresistant tumours and usually, combination chemotherapy is used. There are two major regimens used: High Dose Methotrexate based and Combination therapy with Ifosfamide/Cisplatin/Doxorubicin. The chemotherapy cycles are usually given before surgery as neoadjuvant chemotherapy and after surgery as adjuvant chemotherapy. Both regimens need supportive medications like filgrastim support, antiemeticsetc for decreasing the chances of side effects. In the case of metastatic Osteosarcoma, the initial treatment includes chemotherapy and reassessment. If at reassessment the metastatic lesions (lung) are resectable, then they are removed and chemotherapy is continued.
Ewings sarcoma
Chemotherapy is part of the standard treatment for Ewings sarcoma. They are usually seen in the adolescent and young adult population. After complete staging works up, they are given induction chemotherapy and then taken up for local treatment. The local treatment is either surgery (in case of peripheral lesions) or radiotherapy (pelvic lesions) or both (in case of chest wall Tumours). The total treatment duration is long: 9-12 months as per the treatment protocols. Usually, the treatment is well tolerated by young patients. After treatment, it is very important to be on long term follow up so that any long term treatment-related side effects can be managed promptly. They are highly curative Tumours with proper treatment.
Other bone Tumours
Treatment of chondrosarcoma and most other benign Tumours of bone remains predominantly surgical, although chemotherapy may have a role in dedifferentiated and mesenchymal subtypes. They are usually considered chemoresistant. Incase of low-grade chondrosarcoma, an observation also is an option unless they are symptomatic.
Currently, there is no much role for Targeted therapy in Bone tumours
- Radiation Therapy for Bone Sarcomas
-
Radiotherapy is frequently used in the definitive management of the primary tumour for Ewing sarcoma, but the relative radio-resistance of osteosarcoma and chondrosarcoma means it is only used as definitive treatment when there is no surgical option. These tumours typically require very high doses of radiation (>66Gy) for durable control. Contiguity to sensitive structures, especially at the base of the skull, spine and pelvis, requires high-precision radiotherapy, particularly when total resection seems to be not feasible.
Radiotherapy is not given routinely post operatively, although it may be used in selected high-risk cases or situations such as Chordomas. Radiotherapy also has a palliative role in all tumour types.
- Proton therapy for Bone Sarcomas
-
Protons or carbon ions, often in combination with photons, are increasingly used to treat unresectable primary bone sarcomas.
Excellent outcomes are reported for skull base chondrosarcomas and chordomas in which proton beam radiotherapy combined with surgery can achieve local control rates of approximately 70–90 %. In unresectable or incompletely resectable osteosarcoma the five-year disease-free survival (DFS) was 65 %, and the 5-year overall survival (OS) was 67 %. High local control rates have also been achieved in sacral chordomas.
- Benefits of Proton Therapy for Bone Sarcomas
-
- Higher rates of control rates with Proton therapy compared to IMRT/SRS/Cyberknife. National Cancer database study from the United States has shown the control rates are dramatically better when patients are treated with proton therapy when compared to conventional photon techniques such as IMRT, IGRT, SRS/Cyberknife, etc.
- Low rates of acute and late toxicities. Since higher doses need to be delivered, conventional radiation techniques are associated with a relatively larger burden of side-effects. In fact in certain situations, to limit the probability of side-effects, the radiation dose is reduced. However, Proton therapy ensures the safe delivery of higher doses of radiation therapy with minimal toxicities.
- Reduced risk of Second Cancer. Radiation therapy in combination with chemotherapy is often associated with an increased risk of secondary cancers. Several studies have shown that proton therapy significantly has a lower probability to induce second cancers. This property is especially more useful in younger patients.
- Comparison with IMRT/SRS/Cyber knife
-
VMAT Plan for a Sacral Chordoma
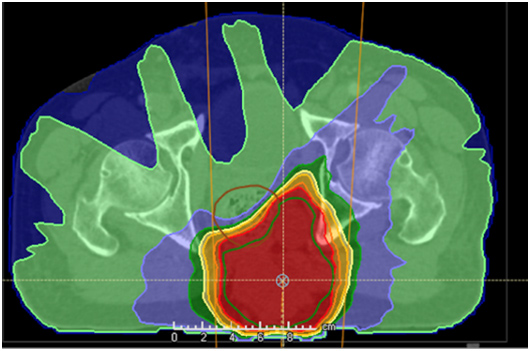
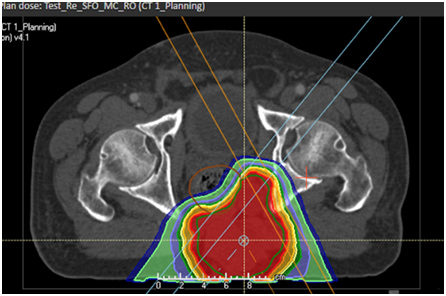
- Proton therapy (IMPT) plan for a sacral Chordoma
-
Demonstrated the sparing on urinary bladder, rectum, pelvic bones and bone marrow.

Other Bone & Soft Tissue Tumours

Copyright © 2023 Apollo Proton Cancer Centre. All Rights Reserved